
Your Target Is Perfect. Your Drug Is Potent. Here's Why It Will Still Fail: The 'Target Fallacy'.
A quantitative framework for understanding why target-based drug discovery fails in translation

A paper from Novartis scientists stopped me mid-scroll. Not because it described a failure — drug development is full of those — but because the failure was so instructive, so precisely illustrative of a trap the entire industry keeps walking into, that it read less like a research article and more like a parable.
The story begins with an elegant scientific plan. It ends with a molecular lesson about the humbling complexity of human biology. And it should change how we think about target selection, translational strategy, and the seductive confidence that comes from a clean in vitro result.
What follows is that story — and three mathematical frameworks that let us reason about it more rigorously.
The Architecture of Confidence
In drug discovery, we build cathedrals on paper. We map pathways, identify nodes, select targets, and construct rationales of extraordinary logical coherence. The best programs feel almost inevitable in their design. Every arrow points in the right direction. Every experiment confirms the hypothesis.
This confidence has occasionally been justified. Gleevec — imatinib — is the canonical example: a single kinase target, BCR-ABL, a single disease defined by a single chromosomal translocation, and a clinical result so dramatic it reshaped oncology. The principle seemed sound: find the right target, block it precisely, and the disease collapses.
But CML is, in retrospect, the exception that proved a much more uncomfortable rule. BCR-ABL is a genuine single point of failure in a cancer that has, for molecular reasons, stripped away most of its redundancy. Most diseases are not like this. Most biology is not like this.
Biological systems did not evolve to be druggable. They evolved to survive. Redundancy, pathway crosstalk, and adaptive rewiring are not bugs — they are the fundamental architecture of resilience.
The assumption that a single, well-chosen target is sufficient to collapse a complex inflammatory or metabolic disease is what I call the Target Fallacy. It is not a foolish assumption — it is a reasonable prior, given some remarkable successes. But it is a prior we have updated too slowly, and the Novartis NEK7 story is the latest evidence of why.
The Plan Was Beautiful
The NLRP3 inflammasome has been one of the most intensively pursued drug targets in inflammation for over a decade. Its activation drives IL-1β and IL-18 release, and dysregulated NLRP3 activity has been implicated in gout, CAPS, atherosclerosis, Alzheimer’s disease, and type 2 diabetes complications.
The Novartis team focused on NEK7, a mitotic kinase that moonlights as an essential scaffold for NLRP3 inflammasome assembly. Critically, NEK7’s role in inflammasome function is independent of its kinase activity — it acts as a structural bridge. The team built a molecular glue degrader, NK7-902, that recruits the CRBN E3 ubiquitin ligase machinery to NEK7, tagging it for proteasomal destruction. NK7-902 was potent, selective, and mechanistically clean, achieving greater than 95% degradation of NEK7 protein.
The preclinical data in mice was compelling. Oral administration strongly degraded NEK7 in splenic tissue and produced meaningful inhibition of IL-1β release in both an acute peritonitis model and a CAPS disease model.
Then came the human and NHP data. In human peripheral blood cells, even with virtually complete NEK7 degradation, IL-1β suppression was partial and highly variable across donors. In non-human primates, sustained 95%+ NEK7 degradation produced only transient and partial inhibition of IL-1β release.
The target was eliminated. The biology barely noticed.
Mathematical Framework I — Pathway Redundancy
The NEK7 result is not mysterious once we model the inflammasome as a network with multiple weighted activation routes, rather than a single linear pathway.
The Core Equation
Define the total NLRP3 activation A as the sum of contributions from all activation routes:
A = Σᵢ wᵢ · aᵢwhere wᵢ is the fractional weight of route i and aᵢ is its activation level (0 to 1). By definition, Σᵢ wᵢ = 1.When we block route j (e.g., by degrading NEK7), the remaining activation is:
A_blocked = Σᵢ≠ⱼ wᵢ · aᵢ = 1 − wⱼ · aⱼThis is the key insight. For NEK7 degradation to suppress NLRP3 activity by ≥80%, we need:
wⱼ ≥ 0.80In mouse cells, NEK7 carries approximately 90% of activation weight — so eliminating it nearly collapses the system. In human and NHP cells, NEK7 appears to carry only ~35% of the weight, with substantial traffic flowing through PTEN-L, mtDNA sensing, and potassium efflux routes. Blocking one road does not close the network.
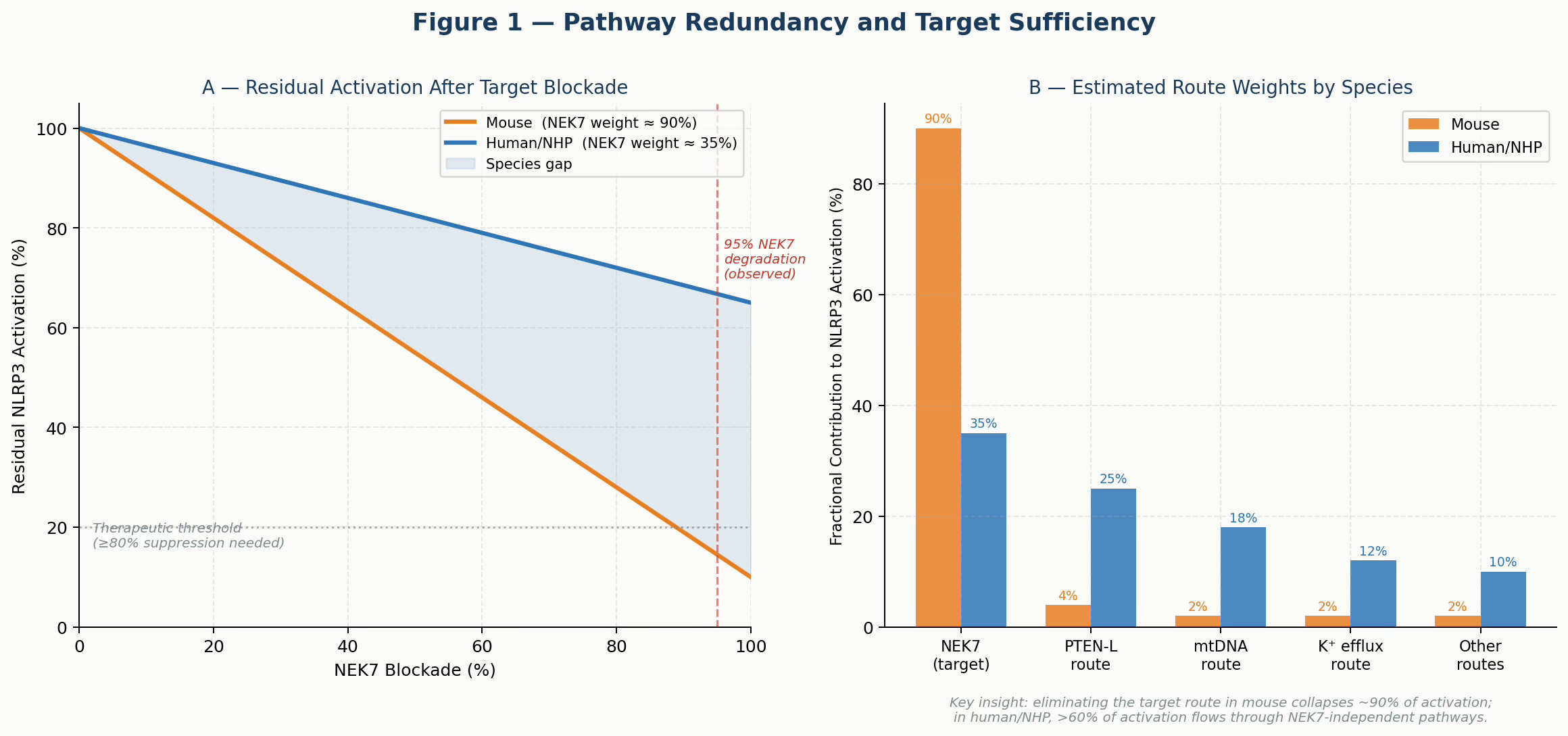
Figure 1. Pathway Redundancy Model. Panel A: Residual NLRP3 activation as a function of NEK7 blockade under mouse vs. human route-weight assumptions. At 95% NEK7 degradation (dashed red line), mouse activation falls below the therapeutic threshold while human/NHP activation remains well above it. Panel B: Estimated fractional contributions of each activation route by species. The NEK7 dominance in mice versus the distributed architecture in humans explains the translational failure.
This framework makes the failure predictable, not mysterious. Any team that had estimated human route weights before committing to lead optimization would have recognized that NEK7 degradation, however complete, was unlikely to produce therapeutic-level pathway suppression in primates.
The practical implication: before selecting a target in a redundant pathway, estimate the target’s fractional contribution in human tissue. This is now possible with modern tools — donor-matched primary cell experiments, siRNA knockdown across a diverse human panel, and pathway flux analysis in organoids. It is not free. But it is far cheaper than a Phase II failure.
Mathematical Framework II — Bayesian Translational Confidence
The NEK7 story is also a story about how evidence should change our beliefs — and about how the current drug development system is structured to update beliefs too slowly, too late.
The Framework
Model our belief in the mechanism as a probability θ — the probability that the mechanism works in humans. We can represent our state of knowledge at each stage as a Beta distribution, Beta(α, β), which is a natural choice for probabilities and updates cleanly as new data arrives.
Prior belief: θ ~ Beta(α₀, β₀)Each experimental stage provides new evidence. After observing s successes and f failures in a relevant species, we update:
Posterior: θ | data ~ Beta(α₀ + s, β₀ + f)The mean of this distribution is α/(α+β), which is our best estimate of P(mechanism works in humans) given all evidence so far.
Applying It to the NEK7 Program
A reasonable prior for a target-based program in inflammation is Beta(3, 5), giving a mean P ≈ 0.375 — consistent with historical Phase II success rates of 30-40% in this space.
Positive mouse efficacy in 4 of 5 models raises the posterior to Beta(7, 6), mean ≈ 0.54. This is the stage where many teams accelerate into IND-enabling studies. The evidence looks convincing.
But then add the NHP pharmacodynamic data: partial and transient IL-1β suppression despite complete target degradation. This is a near-failure — one success out of effectively zero in the most translatable species. The posterior falls to Beta(7, 7), mean ≈ 0.50. Confidence has eroded.
Add the human primary cell variability — inconsistent suppression across donors — and the posterior drops further to Beta(7, 9), mean ≈ 0.44. The belief distribution has shifted materially left. We are now in genuine uncertainty about whether this mechanism is pharmacologically sufficient in humans.
Each experiment is a likelihood update. The question is not whether your compound works in the system you tested. It is whether the full evidence profile justifies conviction in the system that matters — the human patient.
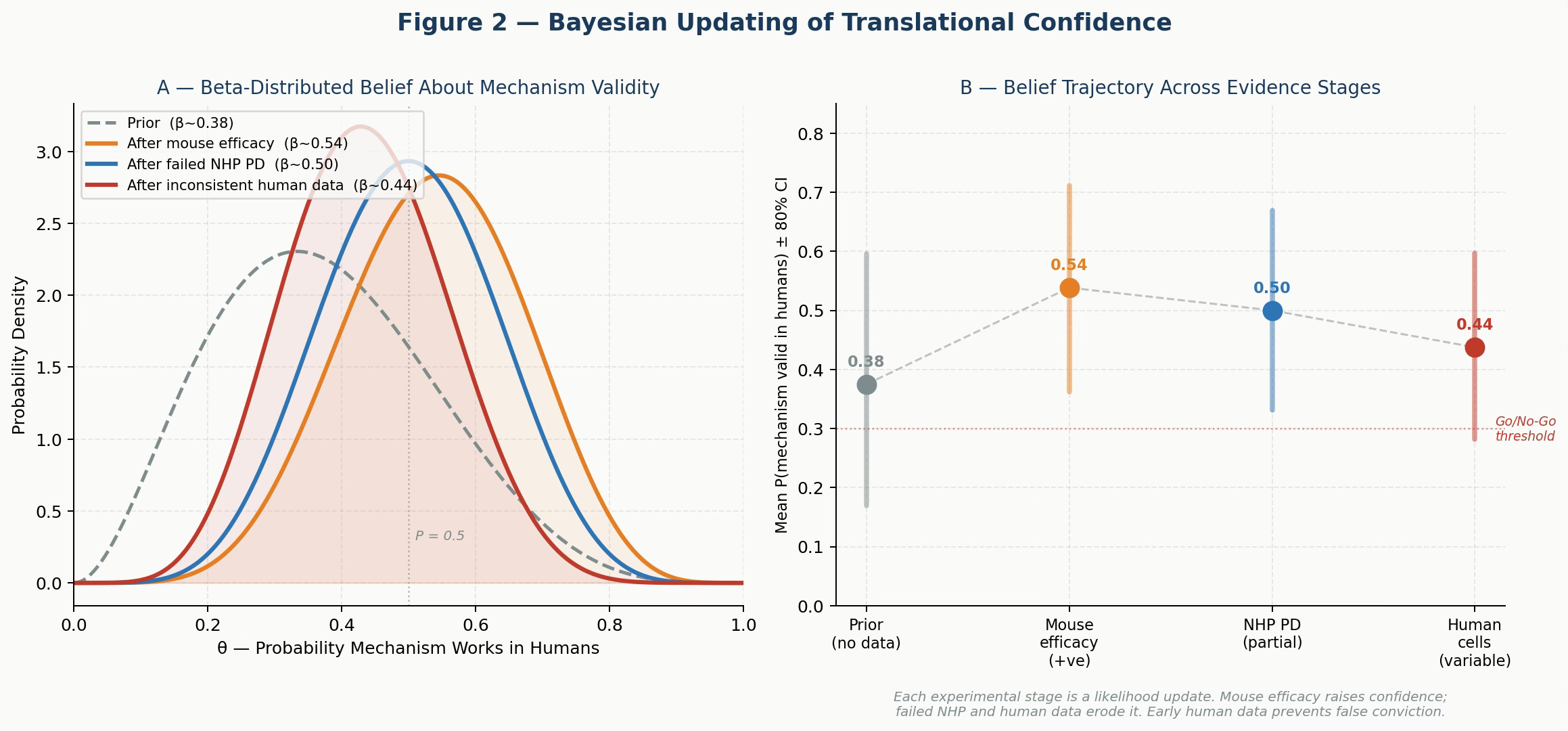
Figure 2. Bayesian Updating of Translational Confidence. Panel A: Beta distributions representing belief in mechanism validity at each evidence stage. Mouse efficacy raises confidence; failed NHP PD and inconsistent human cell data erode it. Panel B: Mean P(mechanism valid) ± 80% credible interval at each stage. The trajectory reveals how mouse-only data produces false conviction that later evidence must correct — often at great expense.
The critical observation from this framework is not the final number. It is the shape of the update trajectory. Programs that incorporate human-relevant data early get the bad news when it is cheap. Programs that defer human data accumulate false confidence, consume resources, and then face a large, late, costly correction.
There is a structural drug-development argument here: the expected cost of generating human primary-cell data in lead optimization is roughly $2-5M for a well-run campaign. The expected cost of a Phase II failure — after the Bayesian update arrives two years later, in the clinic — is $50-200M. The mathematics of belief updating strongly favors early investment in human-relevant evidence.
Mathematical Framework III — Expected Value of Development
The Bayesian framework tells us how evidence should change our beliefs. The expected value framework asks the harder question: given a realistic probability of mechanism validity, which preclinical strategy maximizes the expected return on drug development investment?
The Model
Define the expected value of a program as:
EV = P(mechanism valid) × P(clinical success | mechanism valid) ×
V(approval) − C(preclinical) − P(enter Phase I) ×
[C(Phase I) + P(Phase II) × (C(Phase II) + P(Phase III) × C(Phase III))]where V(approval) is the net present value if the drug reaches market, and each C(stage) represents the expected cost of that stage.
The key variable is P(mechanism valid) — which is precisely what the choice of preclinical strategy determines. A mouse-centric program produces a higher prior estimate of this probability (because mouse models are confirmatory), but a lower true probability. An early human translation program produces a more accurate — and typically lower — estimate, but one that the team can actually rely on.
Why the Arithmetic Favors Early Human Translation
Assume a mechanism with a true P(valid in humans) = 0.20 — typical for a novel inflammatory target. A standard mouse-centric program might estimate this at 0.30-0.40 based on confirmatory rodent data. An early human program would correctly estimate it at or close to 0.20, potentially identifying the translational gap before entering clinical development.
The standard program spends less in preclinical work ($15M vs. $28M with human tissue and NHP PD) but then runs Phase I, Phase II, and potentially Phase III on the basis of a false prior. The early human program front-loads the cost of truth.
The expected value comparison, modeled across a range of mechanism validity probabilities, consistently favors early human translation once the mechanism validity falls below approximately 30-35% — which covers the majority of programs in inflammation, CNS, and metabolic disease.
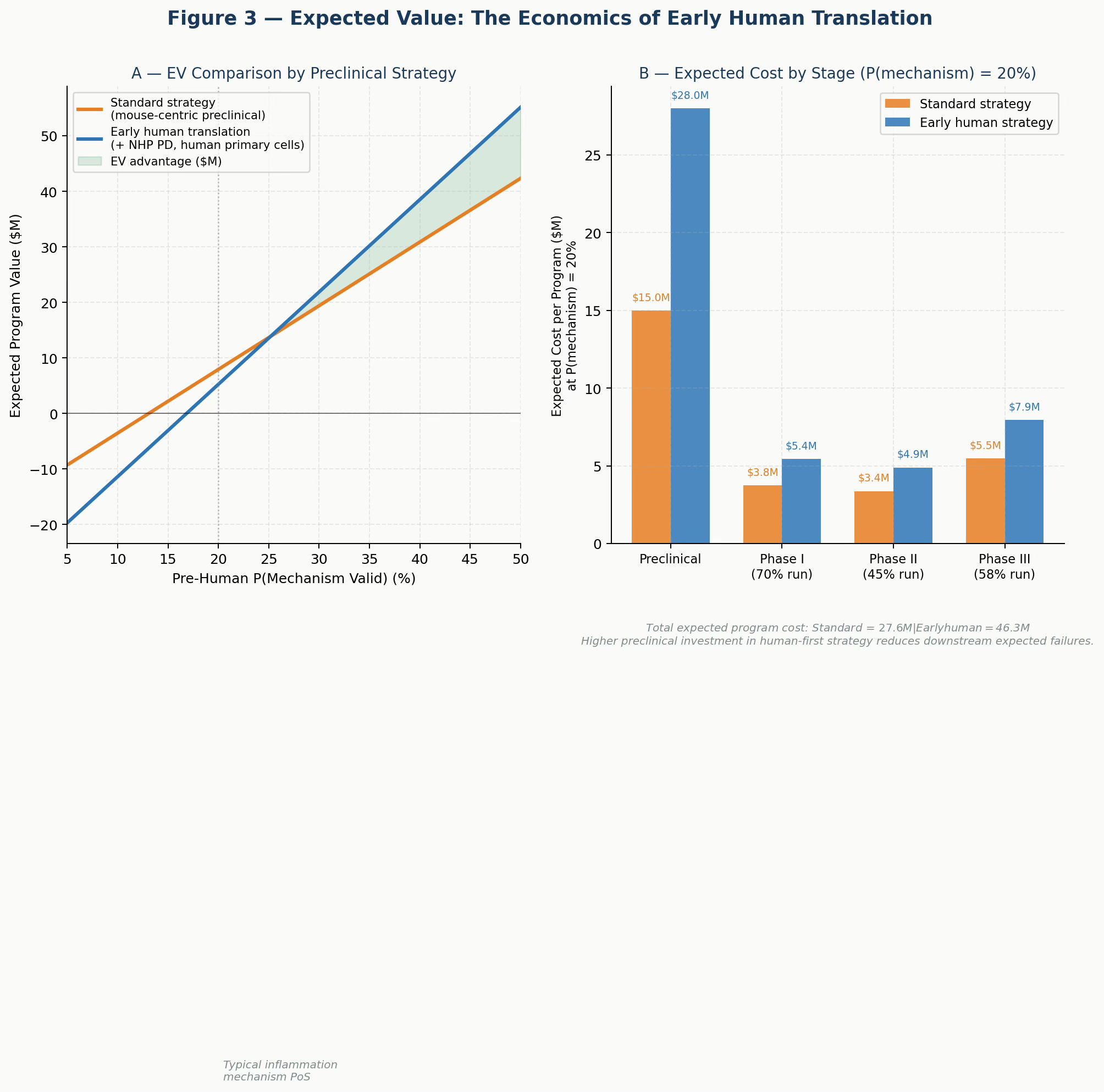
Figure 3. Expected Value of Development by Preclinical Strategy. Panel A: Program EV across a range of pre-human mechanism validity probabilities. The early human strategy (blue) generates higher EV across most of the realistic probability range, with the advantage increasing as mechanism uncertainty rises. Panel B: Expected cost by development stage at P(mechanism) = 20%. Higher preclinical investment in the human-first strategy is more than offset by reduced downstream expected failures.
The cost of false confidence is not paid at the preclinical stage. It is paid in Phase II, when the evidence finally arrives — too late, too expensive, and with patients enrolled.
What Needs to Change
These three frameworks point to the same practical agenda.
First, measure route weights before committing to a target. If a target’s fractional contribution to pathway activation in human primary cells is below 50%, the program faces a high burden to demonstrate therapeutic sufficiency. This is not a reason to abandon the target — it is a reason to investigate polypharmacology, combination strategies, or direct pathway blockade downstream of the redundant nodes.
Second, treat Bayesian updates as decision triggers, not post-hoc observations. Programs should define, in advance, what posterior probability threshold justifies progression to IND-enabling studies. NHP pharmacodynamic failure should move a program into formal go/no-go review, not result in an explanation and a pivot to the next mouse model.
Third, use the EV framework to justify early investment in human translation. The argument that human tissue experiments are “too expensive” for early discovery fails basic expected value arithmetic. The question is never whether early human data costs money. The question is how that cost compares to the expected cost of the alternative.
Fourth, be structurally honest about the limitations of mouse models. A positive result in a murine inflammasome model is evidence that the mechanism is pharmacologically tractable in that species. There is no evidence that the mechanism is relevant in humans. These are different claims, and the organizational habit of treating the former as evidence for the latter has cost the industry billions of dollars and years of patients’ lives.
The Novartis Team Did the Right Thing
The Novartis scientists did not make a mistake. They asked a precise scientific question, built precise tools to answer it, and reported the answer transparently — including the parts that were uncomfortable. That is what science is supposed to do.
The problem is not the science. The problem is an ecosystem that consistently asks the wrong question first: “Can we hit the target?” rather than “Is this target sufficient in humans?” and pays for that inversion at a late and expensive stage.
The NEK7 data suggest that direct NLRP3 inhibitors — which act downstream of the redundant activation routes — may have advantages in human systems. Programs using compounds such as selnoflast and inzomelid, currently in clinical development, will be informative on this point.
But the broader lesson is about first principles. Biological systems are networks, not flowcharts. They evolved to survive perturbation. The three frameworks described here — pathway redundancy, Bayesian belief updating, and expected program value — are not academic exercises. They are practical tools for structuring discovery decisions, ensuring we ask the right questions in the right order.
The most important question in drug discovery is not whether your compound works. It is whether your biology is asking for what your compound offers.
#DrugDiscovery#NLRP3#TranslationalMedicine#InflammasomeBiology#MolecularGlue#PreclinicalModels#TargetFallacy