
How to finance development of a drug that failed commercial assessment
Five models. None complete. One promising direction.

The pharmaceutical industry has a structural problem it rarely states plainly: its entire financing model is built around drugs that do not work too well.
This is not a polemic. It is arithmetic. Venture capital, public equity markets, licensing deals, and royalty finance all depend on recurring revenue. A drug that patients take for years generates an annuity. An annuity can be valued, financed, and sold. The present value of a 20-year biologic prescription is calculable. You can build a company around it.
A drug that cures a disease in a single dose generates one payment. That payment has to recover R&D costs averaging $2 billion per approved asset,¹ cover manufacturing and launch, and yield a return that justifies the original capital. All at once. For a patient population that, in many of the disease areas where curative biology is now most advanced — rare pediatric conditions, autoimmune diseases, inherited metabolic disorders — may number in the hundreds per year.
This is why the most scientifically exciting programs in biopharma are often the least well-financed. The 2024 Deloitte analysis of drugs and biologics launched since 2022 puts average return on R&D investment at 2.5% — one-third of the figure recorded a decade prior.² That decline is concentrated precisely in the categories with the most clinical promise. The misalignment is not incidental. It is intrinsic to how the industry raises and deploys capital.
This piece reviews four financing models that have emerged to address this misalignment, assesses where each falls short, and examines a recently proposed structured finance instrument — cure-backed securities (CBS) — that attempts a synthesis.
The commercial case failure problem
Before reviewing solutions, it is worth being precise about the problem.
Commercial case failure in drug development is not the same as scientific failure. A molecule that clears autoimmune disease in a single infusion has not failed scientifically. It has failed commercially — meaning the projected revenue, discounted across the expected market size and competitive landscape, does not justify the investment required to complete development and launch.
This distinction matters because the remedies are different. Scientific failure requires better biology. Commercial case failure is, in principle, a financing design problem — one that finance has tools to address.
The commercial case problem is worst in three overlapping settings. In orphan diseases, patient populations are small by definition, which caps revenue regardless of pricing. In curative autoimmune therapies — CAR-T for lupus and systemic sclerosis being the most visible current examples — the biology is now showing durable remission in Phase 1 data, but the commercial architecture for one-time curative payment is absent. And in pediatric disease more broadly, pool sizes are small, development timelines are long, and regulatory requirements are appropriately demanding.
In each of these settings, standard venture financing logic does not apply. A Series B biotech investor expects a multiple on invested capital at exit — typically through acquisition or IPO, both of which price a company on projected peak sales. A curative therapy for 400 patients a year, priced at $1.5 million per patient, generates $600 million in peak annual sales. That is not a trivial revenue figure. But it is also not the kind of recurring, predictable stream that commands a large acquisition premium or supports a durable public equity story. The upfront capital required to run a Phase 3 trial in this setting — $300–600 million in many cases — often cannot be recovered under conventional assumptions.
The result is a selection effect: programs in these disease areas are deprioritized, delayed, or abandoned not because the science does not work, but because the financing does not.
Four models and their limits
1. Royalty monetization
Pharmaceutical royalty finance is the oldest structural workaround. Under the standard transaction, a drug developer sells a portion of the future revenue stream from a licensed asset — typically expressed as a percentage of net sales — to a specialized capital provider in exchange for upfront cash. The developer gets capital immediately. The royalty investor takes the revenue risk.
Royalty Pharma, the largest player in this market, has deployed this model at scale across academic institutions, biotechs, and large pharmaceutical companies. DRI Healthcare, over three decades, has acquired 77 royalties on 50 drugs, deploying over $3 billion.³ The Gibson Dunn Royalty Finance Report for 2020–2024 documents sustained expansion of the market, with transaction volume increasing across both traditional royalties (arising from pre-existing license agreements) and synthetic royalties (created specifically for the financing transaction).⁴
The limitation of royalty finance in the curative therapy context is structural. A royalty investor prices a transaction off projected revenue. A curative therapy treating 400 patients a year at $1.5 million per patient generates $600 million in gross annual revenue — but that revenue lasts only as long as the untreated patient pool. Once prevalent cases are treated, incidence-based demand may be 50 or 100 new patients per year. The royalty stream collapses. A royalty buyer modeling a conventional multiple on peak sales gets the arithmetic badly wrong.
Royalty finance also provides capital primarily after proof-of-concept data exist — it is predominantly a late-clinical or post-approval instrument. It does not solve the problem of how to fund Phase 2 and Phase 3 trials in a program with an unconvincing commercial projection.
2. The megafund and research-backed obligations
In 2012, Andrew Lo and colleagues at MIT proposed a more architecturally ambitious solution: a pharmaceutical megafund that would finance a large portfolio of early-stage drug programs and issue tradeable debt instruments — research-backed obligations (RBOs) — collateralized by the portfolio’s intellectual property.⁵
The financial logic behind the megafund is portfolio theory applied to drug development. Individual drug programs have very high failure rates — approximately 95% across all clinical phases for a given indication. But these failures are largely uncorrelated. A molecule targeting lupus fibrosis does not fail because a molecule targeting ALS motor neurons fails. The failure events are biologically, mechanistically, and commercially independent. A sufficiently large portfolio will therefore exhibit predictable aggregate outcomes even if individual program outcomes are highly uncertain. The law of large numbers, applied to drug discovery, converts catastrophic individual failure risk into actuarially manageable portfolio risk.
This is the same logic that underlies insurance and, more directly, asset-backed securities. The megafund would slice that portfolio risk into tranches. Senior RBOs — paid first from the portfolio’s cash flows, carrying low credit risk — would attract institutional fixed-income investors: pension funds, insurance companies, sovereign wealth funds. These investors cannot hold early-stage biotech equity due to volatility, regulatory capital requirements, or mandate restrictions. But they can hold investment-grade debt instruments. Equity tranches, paid last and carrying higher risk, would attract conventional venture capital.
Lo’s simulation work, using historical oncology trial data from 1990 to 2011, found that megafunds of $5–15 billion could yield average equity returns of 8.9–11.4% and bond returns of 5–8% — below venture capital hurdle rates, but within range for pension and insurance investors.⁵ A subsequent paper by Fagnan and colleagues applied the same framework specifically to orphan diseases, where lower development costs and faster FDA timelines improve the math: a $575 million orphan megafund with 10–20 programs could generate double-digit expected returns.⁶
The megafund was never operationalized at scale. The reasons are instructive. Credit rating agencies had no established methodology for scoring a portfolio of preclinical drug candidates — the RBO concept depends on investment-grade ratings to attract institutional debt investors, and those ratings require standardized models that did not exist. Investor education was substantial: the asset class was genuinely new, and institutions with fixed-income mandates were not equipped to diligence biomedical IP. And the concentration of program selection within a single fund manager raised governance questions that had no ready answer.
The megafund’s intellectual contribution — the substitution of portfolio diversification for commercial scale — remains the most rigorous framing of the financing problem. Every serious proposal since has, in some form, borrowed from it.
3. Outcomes-based agreements
Outcomes-based agreements (OBAs) emerged on the payer side as the practical response to a specific tension: a payer asked to pay $1.8 million upfront for a gene therapy whose long-term durability rests on follow-up data spanning three to five years faces an uncomfortable gamble. If the therapy fails at year four, the payer has already paid in full for an outcome that did not materialize.
OBAs restructure this relationship. Under a typical OBA, the manufacturer and payer agree that some portion of the payment is contingent on defined clinical outcomes at defined time points. Bluebird Bio’s commercial launch of Zynteglo (betibeglogene spartacept) for transfusion-dependent beta-thalassemia included a guarantee to refund up to 80% of the therapy’s $2.8 million price if patients did not achieve and maintain transfusion independence within two years.⁷ Novartis launched Zolgensma with an optional five-year instalment plan. Hemgenix, the CSL/UniQure hemophilia B gene therapy approved at $3.5 million — the highest launch price ever recorded for a drug — prompted federal-level discussions about outcomes-based reimbursement architecture.
The most significant recent development in this space is the CMS Cell and Gene Therapy Access Model, which went live in January 2025. Under this model, two gene therapy manufacturers — Bluebird Bio (for Lyfgenia) and Vertex Pharmaceuticals (for Casgevy) — negotiated outcomes-based arrangements with CMS covering Medicaid beneficiaries with sickle cell disease. Thirty-two states, the District of Columbia, and Puerto Rico — collectively representing 84% of Medicaid beneficiaries with the condition — agreed to participate.⁸
OBAs solve a real problem: they lower payer resistance to coverage, align manufacturer revenue with therapeutic durability, and create a financial incentive to collect real-world outcomes data. But they do not address the upstream financing problem. An OBA governs the payment relationship between manufacturer and payer after the drug is approved and launched. It does not fund Phase 2 or Phase 3 trials. A company that cannot convince a venture investor to fund the program in the first place cannot use an OBA as collateral.
There is also an administrative complication that has become more visible as OBAs proliferate: tracking patient outcomes across insurer transitions. American patients change insurers frequently — at job changes, at retirement, at birth of dependents, at state relocation. A five-year outcomes obligation negotiated between a manufacturer and a commercial insurer becomes difficult to enforce if the patient moves to a different insurer in year two. The infrastructure to track, adjudicate, and transfer these obligations does not currently exist in any systematic form.
4. Venture philanthropy
The venture philanthropy model re-routes capital into drug development through disease foundations operating as investment vehicles rather than grant-making organizations. The canonical example is the relationship between the Cystic Fibrosis Foundation (CFF) and Vertex Pharmaceuticals.
Beginning in the late 1990s, the CFF committed approximately $150 million to Vertex’s modulator research program — initially funding work that the venture market considered too early and too uncertain. In return, the Foundation received royalty rights on any products that resulted. The program eventually produced ivacaftor (Kalydeco), then lumacaftor/ivacaftor (Orkambi), and finally elexacaftor/tezacaftor/ivacaftor (Trikafta) — the last of which is eligible for approximately 90% of CF patients by genotype. When the CFF sold its royalty stake to Royalty Pharma in 2014, it received $3.3 billion. A subsequent transaction in 2020 returned an additional $575 million.⁹ That capital has been deployed back into the next generation of CF research.
The CFF model has been replicated, in modified form, in other disease areas. The National Bleeding Disorders Foundation established Pathway to Cures as an explicit venture philanthropy vehicle for hemophilia. NCATS and the Foundation for the NIH operate as federal scaffolding for similar public-private structures, connecting NIH basic research funding with industry development capital. The Bespoke Gene Therapy Consortium, co-led by NIH and FDA with private partners, committed approximately $76 million over five years to rare disease gene therapy programs that would not otherwise attract commercial development capital.
Venture philanthropy has a clear limiting condition: it requires a disease community wealthy enough and organized enough to build an investment-capable foundation, with scientific infrastructure — patient registries, biobanks, natural history data — mature enough to attract initial industry partners. The CF Foundation took decades to build both. Many rare disease communities do not have this foundation. And even for diseases where it exists, the model depends on the foundation absorbing early-stage risk that venture will not — which is only viable when the foundation has sufficient capital to sustain a long development timeline.
The gap none of them closes
Each of these models addresses a different part of the financing problem. Royalty monetization provides capital, but only where adequate commercial scale exists. The megafund solves the diversification problem, but was not operationalized. OBAs align payer incentives but do not fund trials. Venture philanthropy works for a subset of well-organized disease communities.
The gap that remains — the one that explains why curative therapies for rare and autoimmune diseases are systematically underdeveloped — is this: how does a developer finance the clinical development of a therapy that will generate a single payment per patient, in a population measured in hundreds to low thousands per year, when no royalty stream is large enough to monetize, no disease foundation exists, and no OBA helps until after approval?
The conventional answer — price the drug high enough that one payment is sufficient — has produced a sequence of approvals at $1.8 million (Zolgensma), $2.8 million (Zynteglo), and $3.5 million (Hemgenix). Each launch has triggered political, payer, and public backlash that makes subsequent approvals harder. The answer is self-defeating.
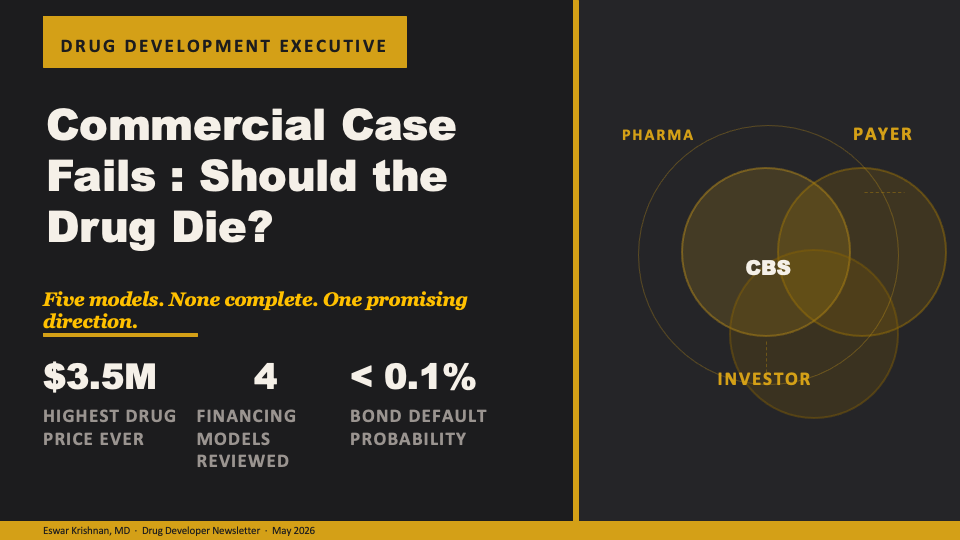
Cure-backed securities: a structured finance proposal
A March 2026 paper in Gene Therapy from Lu, Cherla, Carter, and Mossialos at the London School of Economics proposes an instrument designed specifically to close this gap.¹⁰ Their argument begins with a reframing: the crisis in curative therapy financing is not primarily a pricing crisis. It is a timing crisis.
The manufacturer needs capital now — at development, at approval, at launch. The payer cannot absorb a $1.8–3.5 million charge in a single budget cycle. Both facts are simultaneously true. They are structurally incompatible unless a third party intermediates the time horizon.
Lu and colleagues propose cure-backed securities (CBS): instruments that separate when the payer pays from when the manufacturer receives payment, using the same securitization logic that underwrites 30-year fixed-rate mortgages.
The MBS analogy is precise, not decorative. A mortgage-backed security works because a homebuyer cannot pay $400,000 on day one, a bank does not want to wait 30 years for repayment, and a capital market exists that will buy the future payment stream at a discount. The homebuyer pays monthly over 30 years. The bank sells those payment rights to investors and recovers capital immediately. Investors receive a low-risk yield. The key innovation is the dissociation of who pays, when they pay, and who holds the interim risk.
CBS applies this structure to curative drugs. Under the proposed model, the payer would pay $130,000 per year for up to 30 years — but only while the patient is alive. This is a survival-contingent annuity. No survival, no payment. The manufacturer packages the future payment streams from a pool of treated patients, tranches the pool by actuarial risk, and sells senior and junior bond tranches to institutional investors. The manufacturer receives the net present value of most of that future stream upfront. Critically — and this is the structural departure from all prior models — the manufacturer retains the equity tranche.
The equity tranche is the residual claim: it pays out if the therapy performs better than actuarial expectations and absorbs losses if it performs worse. By retaining this tranche, the manufacturer has a 30-year financial stake in whether the cure holds. This is a genuine incentive alignment that no prior model achieves. OBAs create short-term outcome contingency — two to five years. CBS creates a 30-year financial bond between manufacturer and patient outcome. Patient registries cease to be regulatory overhead and become assets that protect the manufacturer’s equity position.
The authors modeled the structure using Zolgensma case data and ran 1,000 Monte Carlo simulations across base-case, sensitivity, and pessimistic survival assumptions. Senior bond tranches showed default probability below 0.1% under every scenario — investment-grade by any standard. Under pessimistic clinical assumptions, payers paid $1.27 million per patient versus $1.8 million under existing instalment structures or $1.8 million upfront. Manufacturers could front-load 50–83% of expected net present value at time of sale.¹⁰
CBS in context: what it borrows and what it adds
The CBS proposal does not emerge from nowhere. It is the convergence of the four preceding models.
From the megafund, it takes the tranching and pooling logic: diversification across a patient pool reduces the actuarial risk of any individual survival trajectory, just as diversification across drug programs reduces the portfolio failure risk in Lo’s framework. From OBAs, it takes the survival-contingent payment structure — the idea that pharmaceutical payment should track clinical outcome rather than precede it. From royalty finance, it takes the securitization mechanism: future payment rights converted into tradeable instruments sold to institutional capital. And it creates an incentive for real-world outcomes data collection that mirrors the CF Foundation’s registry infrastructure with Vertex.
What CBS adds — the equity tranche retained by the manufacturer — is the piece that prior models lacked. It converts the manufacturer from a party paid once at approval into a party with a continuing financial interest in therapeutic durability across the full patient life.
Limitations and open questions
The Lu et al. paper is careful about scope, and those limits warrant direct acknowledgment.
Orphan disease scope. The CBS model is designed for settings where generic or biosimilar entry over a 30-year horizon is unlikely. Orphan drugs with small patient populations often qualify. For large autoimmune markets — rheumatoid arthritis, psoriasis — where biosimilar entry is certain within 10 years of approval, the annuity structure breaks down. A payer obligated to pay $130,000 per year for 30 years on a therapy for which a $5,000 biosimilar is available in year 12 has been structurally disadvantaged. The CBS structure would need significant modification for non-orphan settings.
Price decoupling. CBS restructures payment timing. It does not constrain what the manufacturer charges. An instrument that spreads a $3.5 million drug over 30 years at $130,000 per year reduces annual budget shock but does not address the underlying pricing question. CBS works best when paired with price negotiation mechanisms — value-based pricing, ICER thresholds, reference pricing — rather than as a substitute for them.
Administrative infrastructure. Tracking patient survival across insurer transitions over 30 years requires infrastructure that does not exist. American patients change insurers at job change, at retirement, at Medicare transition. The payment obligation in a CBS must follow the patient — but no system currently tracks the obligation across payer transitions, adjudicates disputes, or enforces transfer. Building this infrastructure is a non-trivial policy and operational challenge.
Ratings methodology. The megafund failed partly because ratings agencies had no framework to score a portfolio of drug IP. CBS faces an analogous challenge: survival-contingent pharmaceutical annuities are a new asset class. Investment-grade ratings — which the CBS structure depends on to attract pension and insurance capital — require standardized actuarial models that do not yet exist for this instrument type. OBA precedents and the established MBS methodology provide more foundation than existed in 2012, but the gap is real.
The pipeline is coming regardless
CAR-T programs for systemic autoimmune disease — lupus, systemic sclerosis, myositis — are in Phase 1 and Phase 2 now. Several have reported complete drug-free remission in small cohorts. The early data are credible enough that the questions have shifted from “does it work?” to “how durable is it?” and “can it scale?” If any fraction of these programs reaches Phase 3 approval — and the probability is not trivial — the payment infrastructure will face precisely the problem that CBS is designed to address.
The 30-year fixed-rate mortgage did not emerge as a working instrument when the first long-term real estate loan was made. It required decades of financial engineering, regulatory scaffolding, federal mortgage agencies, and secondary market development before it operated reliably at scale. The analogy to curative therapy finance is apt. The intellectual architecture exists. The operational infrastructure does not.
What CBS does — and what the prior models collectively did — is demonstrate that the financing gap for commercially unviable but clinically essential therapies is not a market failure in the pejorative sense. It is a market design failure. The tools to fix it exist. The question is whether the regulatory, institutional, and financial engineering required to implement them can be assembled before the clinical pipeline outpaces the payment system.
References
DiMasi JA, Grabowski HG, Hansen RW. Innovation in the pharmaceutical industry: new estimates of R&D costs. Journal of Health Economics. 2016;47:20–33. doi:10.1016/j.jhealeco.2016.01.012
Deloitte. Measuring the return from pharmaceutical innovation 2024. Deloitte Centre for Health Solutions; 2024. Available at: https://www.deloitte.com/us/en/Industries/life-sciences-health-care/articles/measuring-return-from-pharmaceutical-innovation.html
DRI Healthcare. About DRI Healthcare. Available at: https://drihealthcare.com/about/
Gibson Dunn. Royalty report: royalty finance transactions in the life sciences 2020–2024. 2025. Available at: https://www.gibsondunn.com/royalty-report-royalty-finance-transactions-in-the-life-sciences-2020-2024/
Fernandez J-M, Stein RM, Lo AW. Commercializing biomedical research through securitization techniques. Nature Biotechnology. 2012;30(10):964–975. doi:10.1038/nbt.2374
Fagnan DE, Gromatzky AA, Stein RM, Fernandez J-M, Lo AW. Financing drug discovery for orphan diseases. Drug Discovery Today. 2014;19(5):533–538. doi:10.1016/j.drudis.2013.11.007
Bluebird Bio. Bluebird bio announces U.S. commercial infrastructure to enable patient access to ZYNTEGLO. Business Wire. August 17, 2022. Available at: https://investor.bluebirdbio.com/news-releases/news-release-details/bluebird-bio-announces-us-commercial-infrastructure-enable
Centers for Medicare & Medicaid Services. Cell and Gene Therapy Access Model — frequently asked questions. 2025. Available at: https://www.cms.gov/cgt-access-model-frequently-asked-questions
Cystic Fibrosis Foundation. Cystic Fibrosis Foundation receives $3.3 billion royalty pay out. Philanthropy News Digest. 2014. Available at: https://philanthropynewsdigest.org/news/cystic-fibrosis-foundation-receives-3.3-billion-royalty-pay-out
Lu JM, Cherla AJ, Carter AW, Mossialos EA. Securitization as a means to pay for cell and gene therapies for orphan diseases: a simulation study. Gene Therapy. 2026. doi:10.1038/s41434-026-00604-6
Author: Eswar Krishnan, MD Date: 2026-05-09
Eswar Krishnan is a physician and principal at Olmsted Capital LLC. He consults on clinical development strategy at Drug Development Associates.
#DrugDevelopment #RareDisease #BioPharmFinance #StructuredFinance