
Block One Road. Biology Takes Another
We Killed the Target. The Disease Didn't Care

A paper from #Novartis scientists stopped me mid-scroll. Not because it described a failure — drug development is full of those — but because the failure was so precisely illustrative of a trap the entire industry keeps walking into that it read less like a research article and more like a parable.
The story begins with an elegant scientific plan. It ends with a biological lesson about redundancy, evolution, and the uncomfortable gap between species. And it should change how we think about target selection, translational strategy, and the seductive confidence that comes from a clean result in a mouse.
The Architecture of Confidence
In drug discovery, we build cathedrals on paper. We map pathways, identify nodes, select targets, and construct rationales of extraordinary logical coherence. The best programs feel almost inevitable in their design. Every arrow points in the right direction. Every experiment confirms the hypothesis.
This confidence has occasionally been justified. Gleevec — imatinib — is the canonical example: a single kinase target, BCR-ABL, a single disease defined by a single chromosomal translocation, and a clinical result so dramatic it reshaped oncology. Find the right target, block it precisely, and the disease collapses. The principle seemed sound.
But CML is, in retrospect, the exception that established a flawed template. BCR-ABL is a genuine single point of failure in a cancer that has, for molecular reasons, stripped away most of its biological redundancy. Most diseases are not like this. Most biology is not like this.
Biological systems did not evolve to be druggable. They evolved to survive. Redundancy, pathway crosstalk, and adaptive rewiring are not bugs — they are the fundamental architecture of resilience.
The assumption that a single, well-chosen target is sufficient to collapse a complex inflammatory or metabolic disease is what I call the Target Fallacy. It is not a foolish assumption — it is a reasonable one, given some remarkable successes. But it is an assumption we have updated too slowly, and at extraordinary cost.
The Plan Was Beautiful
The NLRP3 inflammasome has been one of the most intensively pursued drug targets in inflammation for over a decade. Its activation drives the release of interleukin-1β and IL-18 — inflammatory signals implicated in gout, rheumatoid arthritis, cardiovascular disease, Alzheimer’s disease, and a spectrum of autoinflammatory conditions. Getting control of NLRP3 activation has been a central ambition of the inflammatory disease field.
The Novartis team focused on NEK7, a mitotic kinase that serves as an essential structural scaffold for NLRP3 inflammasome assembly. The key insight was that NEK7’s role in the inflammasome is not catalytic — it does not drive a chemical reaction — but architectural. It holds the complex together. This means that rather than trying to block an enzymatic activity, you could simply remove the protein entirely.
The drug they built to do this — NK7-902 — was a molecular glue degrader. It works by tricking the cell’s own disposal machinery into destroying NEK7, tagging it for proteasomal degradation via the CRBN E3 ubiquitin ligase system. NK7-902 was potent, selective, and mechanistically elegant. In biochemical assays, it worked exactly as designed: it degraded more than 95% of NEK7 protein.
On paper, eliminating the scaffold should collapse the inflammasome. The logic is sound. The chemistry is elegant. The execution was flawless. And in mice, it worked.
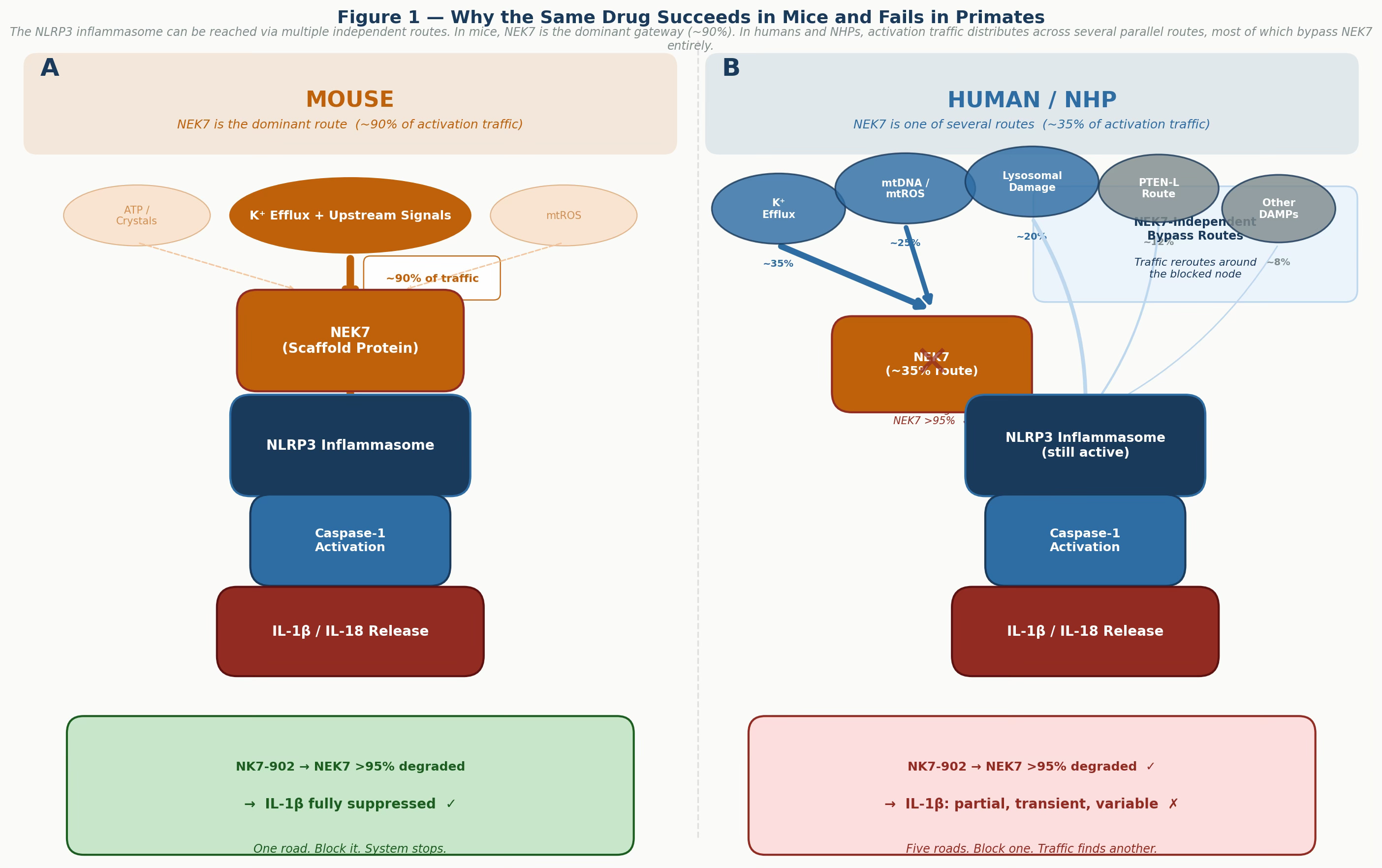
Figure 1 — Why the same drug succeeds in mice and fails in primates. Panel A: in mouse cells, NEK7 is the dominant gateway for NLRP3 activation, carrying roughly 90% of activation traffic. Blocking it effectively shuts down the system. Panel B: in human and NHP cells, activation traffic distributes across multiple parallel routes, most of which bypass NEK7 entirely. Eliminating one node reroutes — it does not stop — the pathway. Note: route weight estimates are conceptual and illustrative of the biological principle.
The Biology Disagreed
The preclinical data in mice was encouraging. Oral NK7-902 strongly degraded NEK7 in splenic tissue and produced meaningful inhibition of IL-1β release in both an acute peritonitis model and a CAPS disease model. The team had a molecule that worked, a target that was tractable, and a disease rationale that held in rodents. Any discovery team would have been cautiously optimistic.
Then came the human and non-human primate data, and the story changed entirely.
In human peripheral blood cells, even with virtually complete NEK7 degradation, IL-1β suppression was partial and highly variable — differing substantially across individual donors and experimental conditions. That variability is itself a signal. When a mechanistically clean intervention produces inconsistent functional effects across human donors, the system is communicating something important about its internal architecture.
In non-human primates, the dissociation was even more striking. Oral dosing at 2 mg/kg produced sustained NEK7 degradation exceeding 95% in blood — the compound had done exactly what it was designed to do. IL-1β inhibition was transient and partial. The target was eliminated. The biology barely noticed.
The target was eliminated. The biology barely noticed. This is a sentence that should be framed and hung in every drug discovery department in the world.
What this tells us is not that NEK7 is unimportant in inflammasome biology. In mouse cells, it clearly is important — essential, even. What it tells us is that the inflammasome pathway in primates has evolved redundant activation routes that do not depend on NEK7. When one road is blocked, the system takes a different highway. Evolution, unlike our target-validation assays, tested this over millions of years.
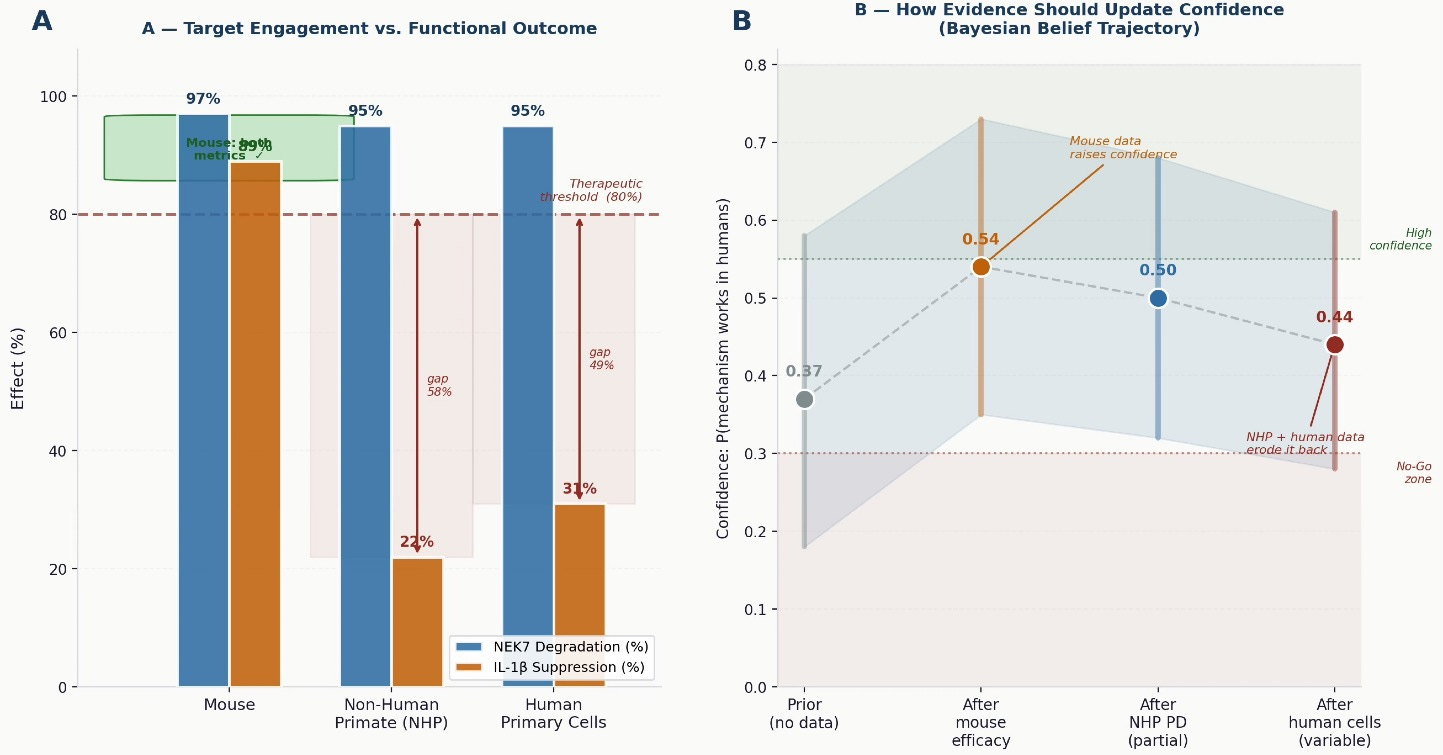
Figure 2 — The efficacy disconnect and what the evidence should tell us. Left: NK7-902 achieves near-identical NEK7 degradation across all three species. IL-1β suppression tracks with species — not with compound performance. Right: how confidence in the mechanism should update as each piece of evidence arrives. Mouse efficacy raises it; NHP and human data erode it. Programs that collect human-relevant data early get the correction cheaply. Programs that defer it pay compound interest.
Why This Keeps Happening
The NEK7 story is not unique. It sits in a long and expensive tradition. The p38 MAPK inhibitors showed exceptional preclinical promise in rheumatoid arthritis for nearly a decade before a string of clinical failures revealed that the human RA synovium is far more pathway-redundant than any rodent model had suggested. The CANTOS trial demonstrated that blocking IL-1β with canakinumab reduces cardiovascular events — the biology was real — but the increase in fatal infections and the commercial calculus made it unworkable. The list of programs that succeeded in mice and failed in humans is long enough to be its own clinical speciality.
There is a structural reason this pattern persists. Mouse models are inexpensive, fast, and genetically manipulable. They generate clean, interpretable data. They are scientifically satisfying in a way that messy human primary cell experiments rarely are. The incentive architecture of early drug discovery — publication pressure, milestone timelines, the cost differential between rodent and human studies — consistently pushes teams toward the models most likely to produce affirmative results, not the models most likely to predict human outcomes.
We select for confidence. Biology rewards robustness.
The two are not the same thing, and the gap between them is where drug development money goes to disappear.
The Species Gap Is a Biological Fact, Not a Technical Problem
A common response to translational failures is to reach for better tools: humanized mice, organoids, organs-on-chips. These are genuinely valuable, and investment in them is warranted. But there is a more fundamental issue that better experimental systems alone cannot resolve.
Human and mouse immune systems diverged roughly 65 to 80 million years ago and have been under substantially different selective pressures ever since. The human NLRP3 pathway appears to have developed redundant NEK7-independent activation routes that are simply not present in the mouse — not because our models are imperfect, but because that is what evolution produced. This is not a gap in our understanding that better technology will close. It is a biological reality that demands a different strategic response.
The strategic response is to ask the critical question earlier. Not “does our target matter in this pathway?” but “is this pathway, in human tissue, sufficiently non-redundant that eliminating our target will produce the functional outcome we need?” These are different questions. The second one requires human data. And it needs to be asked before lead optimisation is complete — not at Phase II.
The question is not whether your target matters. The question is whether, in human biology, your target is a single point of failure — or merely one of several roads to the same destination.
The Cost of Getting This Wrong
The financial argument for earlier human translational work is less intuitive than it should be. Adding NHP pharmacodynamic studies and human tissue experiments in early discovery can double or triple the cost of lead optimisation. Program teams face pressure to move fast, hit milestones, and advance compounds. Spending more time and money preclinically feels like slowing down.
But the arithmetic is straightforward. A Phase II failure in an inflammatory indication costs between $50 million and $200 million, depending on trial size and duration. A Phase III failure costs an order of magnitude more. The industry-wide failure rate for drugs entering Phase II in inflammation and immunology is approximately 70%. If even a fraction of those failures trace back to a translational assumption that human primary cell data would have challenged — and the evidence suggests many do — the financial case for front-loading that investment is overwhelming.
There is also a patient cost that rarely enters these calculations directly. Every program that reaches clinical trials based on mouse efficacy data represents a commitment of patient time, risk, and hope. Phase II failures are not just expensive — they are a burden on people who enrolled because they or their physicians believed the mechanism had a reasonable chance of working. Better preclinical translation does not just protect pipeline value. It protects patients.
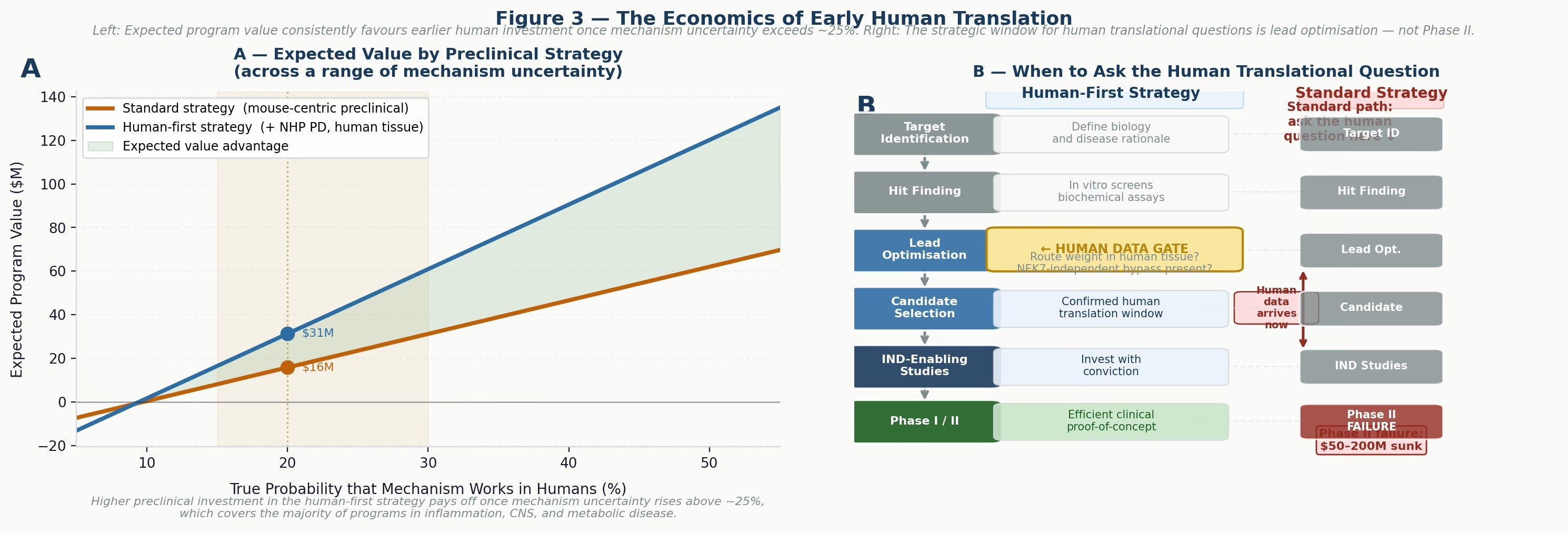
Figure 3 — The economics of early human translation. Left: expected program value as a function of true mechanism probability in humans. The human-first strategy consistently outperforms once mechanism uncertainty rises above roughly 25%, which covers the majority of programs in inflammation, CNS, and metabolic disease. Right: the strategic decision timeline — the window to ask the human translational question is lead optimisation, not Phase II. Waiting until clinical studies to discover pathway redundancy is the most expensive form of discovery research.
What Needs to Change
The path forward is not a single intervention. It is a reorientation of how discovery programs are structured, and what counts as evidence of confidence versus evidence of translatability.
Human primary cell data should function as a gate, not a late confirmatory box to tick. When a compound shows robust efficacy in mouse models, the immediate question should not be which additional mouse models to run, but what happens in human primary cells and tissue. Variability across donors — as seen in the Novartis data — should be interpreted as information about the mechanism, not experimental noise to be averaged away.
NHP pharmacodynamic studies, where feasible, should be incorporated before lead optimisation is complete. This is a significant resource commitment, and not every program will justify it. But for mechanisms where the translational risk is structurally high — innate immune pathways, CNS targets, metabolic nodes — the investment is warranted. A partial NHP pharmacodynamic result, collected early, is worth far more than a complete one collected at IND.
Phenotypic approaches deserve serious investment alongside target-based programs. Phenotypic screens measure functional outcomes in human or humanised systems rather than target engagement alone. A compound that suppresses IL-1β release in human peripheral blood across a diverse donor panel — through whatever mechanism — has demonstrated human-relevant functional activity that no target-engagement assay can match. This is not an argument against target-based discovery. It is an argument for complementing it.
Finally, we need institutional honesty about what mouse efficacy data actually proves. A positive result in a murine model is evidence that the mechanism is pharmacologically tractable in that species. It is not evidence that the mechanism is sufficient in humans. Treating the former as evidence for the latter — routinely, under timeline and resource pressure — is the Target Fallacy in its most common and costly form.
What the NEK7 Story Really Tells Us
The Novartis scientists did not make a mistake. They asked a precise scientific question, built precise tools to answer it, and reported the answer honestly — including the parts that were uncomfortable. The paper is a model of scientific integrity. That the result was not what they hoped for is not a failure of the team. It is a failure of the assumptions that the field, as a whole, brought to the program.
The problem is not the science. The problem is an ecosystem that consistently asks the wrong question first — can we hit the target? — rather than the more important one: is this target sufficient in humans? And pays for that inversion at a late, expensive, and patient-facing stage.
The NEK7 data also points forward constructively. Direct NLRP3 inhibitors — which block the protein itself rather than an upstream activator — may have advantages in human systems precisely because they sit downstream of the redundant activation routes that bypass NEK7. Compounds like selnoflast and inzomelid, currently in clinical development, will be informative on exactly this point. The biology is telling us where to look next.
But the broader lesson is about first principles. Biology is a network, evolved over hundreds of millions of years to survive perturbation. Blocking one node rarely collapses it. Before we invest in any mechanism at scale, we owe it to patients — and to the basic integrity of the enterprise — to find out whether we are blocking a single point of failure, or merely rerouting traffic.
The most important question in drug discovery is not whether your compound works. It is whether your biology is asking for what your compound offers.
The Target Fallacy will not be the last story of this kind. But if we take it seriously — if we use it to restructure how and when we ask translational questions — it might be one of the last times we are surprised by it.
#DrugDiscovery #NLRP3 #TranslationalMedicine #TargetFallacy #PreclinicalModels #InflammasomeBiology #NEK7 #Novartis